(感謝林鈺杰助教製作)
一、
N2
分子為單、雙或三鍵?
1. 分別建立N2、N2H2、N2H4分子結構模型,置入超晶胞後進行結構最佳化。
2.
對三種分子結構進行population analysis,計算其bond order。
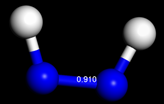
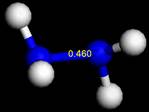
3.
三種分子的bond order分別為1.33、0.91、0.46,由於bond order比例接近3:2:1,因此判定N2之鍵級為三鍵。

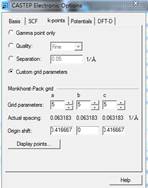
二、以加 Hubbard U 方法將 NiO 之 Ni 的佔據與未佔據 d 軌域能量拉開至 1.5 eV。
1.
從Material Studio中import NiO之晶體結構,並對其進行結構最佳化。
2.
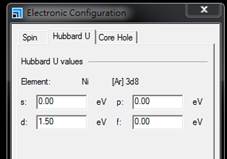
從結構中選取Ni原子並從Modify => Electronic
Configuration中調整Hubbard U之值。
![]()
3.
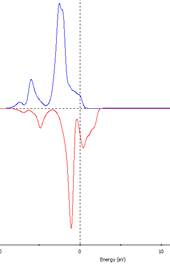
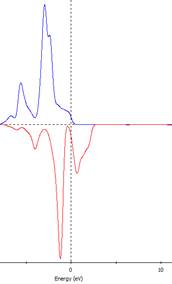
測試不同U值直到PDOS中Ni之佔據與未佔據 d 軌域能量拉開至 1.5 eV。U值約為1.0
eV時可達成。
U=1.0 eV
U=1.5 eV
![]()
![]()
三、預測體心鐵的自發磁化率。
1.
參考課本p445
四、呈現
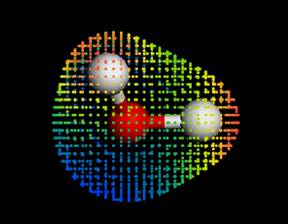
H2O 分子的極性分佈。
1.
建立H2O分子結構模型,置入超晶胞後進行結構最佳化。
2.
進行能量計算並勾選electron
density difference選項。
3.
畫出電荷密度及potential分布並在電荷密度的isosurface中選取color
by mapped field可畫出極性分布。
五、從能帶結構及總能量觀點,說明一維氫原子鏈不穩定。
1.
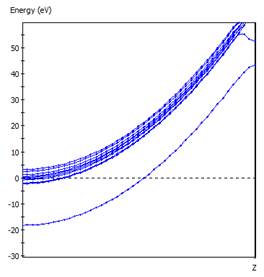
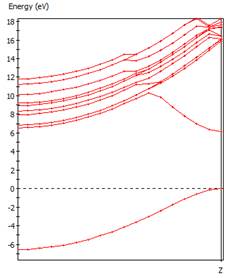
首先建立氫分子模型以及一維氫原子鏈,氫原子鏈模型可藉由調整單一方向晶胞長度至氫分子鍵長而產生。


2.
進行總能及能帶結構計算,可得知形成氫分子相對穩定,請參考課本之解釋。(Peierls Distorsion)
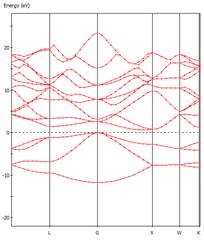
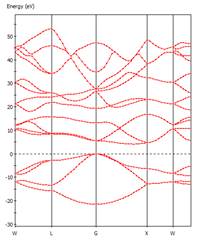
六、矽與鑽石能隙大小各為何?直接或間接?畫出其導帶底部之出單 k 點波函數(密度分佈)。
1.
從Material Studio中import Silicon及diamond之晶體結構,轉換為primitive cell後進行結構最佳化以及能帶結構計算,
可得兩者皆為間接能隙,能隙值為0.61
eV (Si)及4.14 eV(Diamond)。
Si
Diamond
2.
找尋其導帶最低點之k點,並將origin shift至該點進行DOS計算,以DOS計算所得之可畫出單一k點之波函數。
![]()
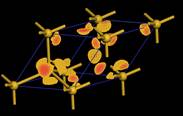 Si
Si 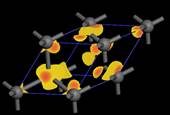 Diamond
Diamond
七、反鐵磁態的 MnO 其同一磁距面(即同為向下或同為向上)是沿
111 面方向交替出現,試建構此初始態之並取最小晶胞。
1.
參考課本p456-p467
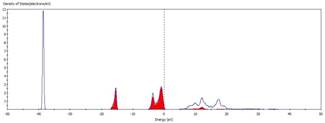
八、MgO的價帶頂部主要由哪種元素所貢獻?
1.
從Material Studio中import MgO之晶體結構,並對其進行結構最佳化。
2.
計算Density
of state並分別畫出TDOS、Mg LDOS以及O
LDOS,將Mg及O的LDOS分別貼到TDOS中可看出兩者在TDOS中之貢獻,因此可判斷價帶上緣由O貢獻。
Mg-LDOS
in TDOS
O-LDOS in TDOS